
How You Look at a Protein Can Change Its Structure
The most common way to get a picture of the atomic structures of a protein might actually cause a change in the protein’s structure, a team of researchers discovered.
X-ray crystallography has been used to examine protein structures since the 1950s and remains the most popular method among researchers in the field. It’s a process that includes multiple steps, including forcing the molecules into a crystalline structure and cooling the protein to 77 degrees kelvin (-321 degrees Fahrenheit). Another commonly used experimental method, nuclear magnetic resonance (NMR) spectroscopy, also obtains atomic-level detail of protein structure, but by a very different method: solution-based samples are placed in a large magnet that sends magnetic field lines through the solution. Because the protein remains in a liquid solution at room temperature, many researchers consider the protein to be in a more natural state than x-ray crystal structures.
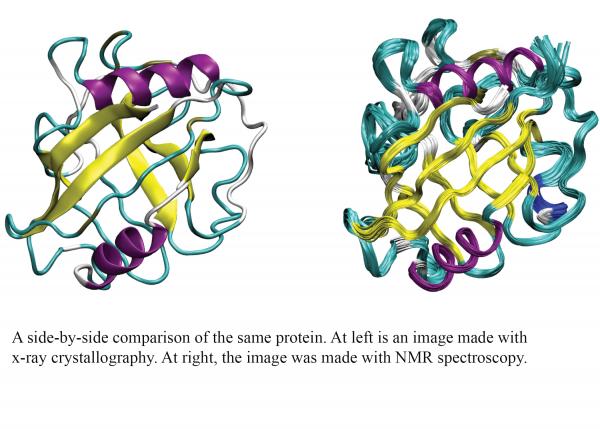 To find out whether these two experimental methods reveal the same structures for a given protein, researchers in the lab of Corey O’Hern, professor of mechanical engineering & materials science, looked at images of the same proteins taken using x-ray crystallography compared to those taken using NMR spectroscopy. The results were recently published in Proteins: Structure, Function, and Bioinformatics.
To find out whether these two experimental methods reveal the same structures for a given protein, researchers in the lab of Corey O’Hern, professor of mechanical engineering & materials science, looked at images of the same proteins taken using x-ray crystallography compared to those taken using NMR spectroscopy. The results were recently published in Proteins: Structure, Function, and Bioinformatics.
“We found fundamental differences - very slight ones, but we think they give important insight about not just the experimental techniques, but the protein structure itself,” said John Treado, a Ph.D. student in O’Hern’s lab, and lead author of the study.
One of the specific differences was that the images created using NMR spectroscopy showed that the amino acids were packed slightly denser in the protein cores than those created using x-ray crystallography.
“The amino acids in x-ray crystal structures at cryogenic temperatures do not move significantly and become stuck in less well-packed states,” Treado said. “But the NMR structures, because they’re in solution at room temperature, the structures fluctuate more, and the core amino acids can find states that are a little bit denser.”
To test this hypothesis, they created a computer simulation in which they put amino acid-shaped particles inside a box and compressed it to determine how densely they could pack the particles. When they included thermal fluctuations to simulate the conditions of NMR spectroscopy, the researchers found that the particles can pack more densely than that found for x-ray crystal structures.
“And if we let the amino acids move around more, they can pack more densely, and the structures will look more like NMR structures,” he said. “So our conclusion is that NMR structures are denser because the amino acids are allowed to undergo thermal fluctuations at room temperature.”
Most likely, these differences come down to one of two possible causes. One points to the very cold temperatures used in x-ray crystallography. “In that case, we should figure out ways to crystallize proteins at high temperatures.” But another possibility is that it’s the crystalline structure itself that’s restricting the fluctuations of the protein.
“If that’s the case, we should really caution people away from using crystal structures and encourage additional studies to generate NMR structures,” he said.
The field of protein design holds great promise for the development of new materials, catalysts, drugs, and drug delivery systems. To fully tap that potential, though, researchers need a better understanding of protein structures.
“If I'm in a pharmaceutical company and I want to understand how a drug binds to a protein, I’ll need to know the variability in the structure,” Treado said. “For example, will this drug always be able to bind or will there be some structural variations that do not allow binding?”