
Researchers Solve Problem of Reassembling Protein Puzzles
The field of protein design holds great promise for the creation of new materials, catalysts, drugs, and drug delivery systems. To fully tap that potential, though, researchers need to perfect their understanding of existing structures – to the point of being able to take apart natural proteins and correctly reassemble them.
 To that end, a team of researchers at Yale has developed a computational method that allows them to successfully predict how the amino acids of a deconstructed protein would fit back into place. Key to this was treating the protein like a 3-dimensional jigsaw puzzle. The results of the research, led by Prof. Corey O'Hern and Prof. Lynne Regan, were recently published in Protein Engineering, Design, and Selection, which also featured the study on its cover.
To that end, a team of researchers at Yale has developed a computational method that allows them to successfully predict how the amino acids of a deconstructed protein would fit back into place. Key to this was treating the protein like a 3-dimensional jigsaw puzzle. The results of the research, led by Prof. Corey O'Hern and Prof. Lynne Regan, were recently published in Protein Engineering, Design, and Selection, which also featured the study on its cover.
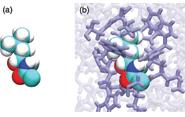
“It’s as if a toy company took the amino acids out of the core of the structure and left the exterior as the scaffold,” said O’Hern, whose primary appointment at Yale is in Mechanical Engineering & Materials Science. “Then they give you the amino acids as 3D puzzle pieces, and say ‘put the puzzle back together.’ That’s what we did.”
The researchers developed techniques to model amino acids as building blocks with complex geometrical shapes based on protein crystal structures that have been collected over the past 30 years in the Protein Data Bank, an online database of protein structures. With the new modeling tools, the researchers can try all the possible ways that amino acids can fit into the cores of protein structures. “We showed that there is only one way that amino acids in the core of a protein can fit together, and we found it for each protein in the database,” he said.
The groundbreaking aspect of the Yale researchers’ work is the simplicity of the geometry-based approach. It’s in stark contrast to two other common approaches for predicting protein structure: One focuses only on analyzing what is found in natural proteins, using an ad hoc ‘knowledge-based’ energy function. The other includes in the energy function multiple types of physical interactions, such as electrostatics, Van der Waals, and hydrogen bonding. The success of O’Hern and Regan’s approach illustrates the dominance of steric repulsion – that is, repulsive interactions that prevent atoms from overlapping each other – in specifying the structure of protein cores.
O’Hern noted that protein design is like any other kind of engineering. “You can’t design something like a new car, if you don’t know what the old cars are like,” he said. “You need to know what worked in the existing functional models, and then you can take the existing designs, and improve them.”
As for using this approach to design new proteins, O’Hern said the first efforts will likely focus on mutations. “What we can do now is say, ‘Well we don’t want the natural occurring amino acid in a particular location, we want to improve the structure with a mutated sequence. So what’s the best alternative amino acid that can fit in a region formerly occupied by the wild type amino acid?’”
Other co-authors on the paper included graduate students Diego Caballero and Alejandro Virrueta.